Chromatin structure and the regulation of gene expression
The specialized cells of a complex organism express only a characteristic subset of genes, although the vast majority of cells contain the same and complete genome. To make a prediction from the primary DNA sequence whether and to which extent a certain gene is transcribed or not is impossible. In the nucleus the DNA becomes highly compacted by DNA interacting proteins forming the chromatin. Modifications of the degree of chromatin compaction play an important role in the regulation of gene expression. The most direct modification of DNA that does not change its primary sequence, which can be heritable and is reversible, is DNA methylation. The adding of methyl groups to DNA is commonly directed to carbon number 5 in the pyrimidine ring of cytosine (5meC). DNA methylation is catalyzed by DNA methyltransferases (DNMTs) and occurs frequently at symmetric CpG motifs in mammals, and additionally at asymmetric motifs in plants (CpNpG and CpHpH). De novo DNMTs newly methylate cytosines of DNA and are predominantly expressed during early embryonic development, whereas semi-conservative maintenance DNMTs transmit methyl groups to hemimethylated DNA that origins from DNA replication. DNA methylation typically is associated with transcriptional repression possibly by direct blocking of transcription factor binding or by recruitment of histone deacetylases, thus impeding the decondensation of higher order conformations.
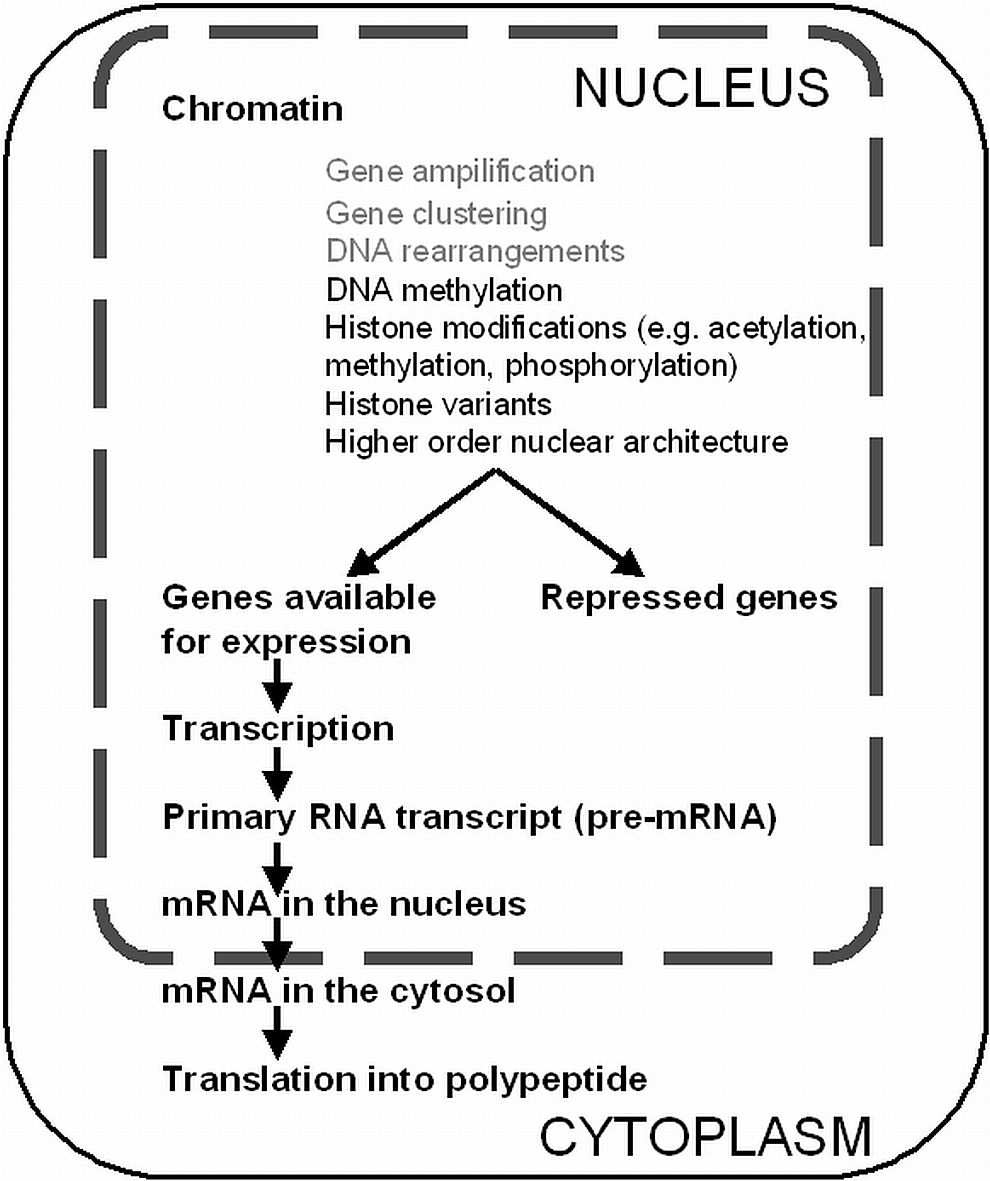
The first level of chromatin compaction is the 10 nm fiber, in which the DNA is wrapped around nucleosomes, an octamer composed of the four histone protein types H2A, H2B, H3 and H4. This 10 nm fiber becomes further compacted by the interaction with linker histone H1 and other proteins. Posttranslational modifications (PTMs) at the N-termini of all histones can alter the degree of chromatin compaction. In vivo observations suggest that transcriptionally silent genes mostly occur within domains of condensed heterochromatin, whereas in many cases actively transcribed genes are found within domains of less condensed chromatin.
Histone modifications occur in a dynamic fashion. Through altering the degree of chromatin compaction, PTMs of histone tails create chromatin structures favorable either for activation or repression of genes, depending on the genomic context and the combination of modifications at a given site. Hereby two basic mechanistic principles have been recognized: The introduction of PTMs can create binding sites for the recruitment of non-histone effector proteins, or it breaks up nucleosomal interactions leading to chromatin decompaction. Modifications found at distinct residues of the histone protein N-termini include - among others - lysine acetylation, lysine and arginine methylation as well as serine and threonine phosphorylation. Various enymes have been identified that specifically introduce or remove these modifications. Such enzymes include histone acetyltransferases (HATs) and deacetylases (HDACs), histone lysine methyltransferases (KMTs) and histone lysine demethylases (KDMs).
RESEARCH FOCUS IN OUR LAB
We analyze the biological relevance of histone H3 variants, their post-translational modifications as well as their specific assembly into chromatin.
We intend to elucidate, how epigenetic decisions contribute to the selective transregulation of the host genome in hepatocytes in the course of infantile and adult chronic Hepatitis B virus infection.
We are developing anti-hepatitis-B-viral gene therapy strategies using the stable, episomally replicating pEPI-1/pMARS vector system.
| 


